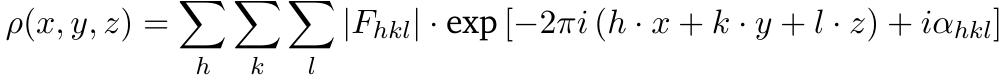This is going to be a very Rosetta specific article, if you haven’t used it before this probably is not for you. In short: I recently had to do a lot of simulations in Rosetta, ranging from simple relaxations over protein design to comparative modelling. As the problem with simulations in Rosetta is mostly to make it run I usually do not add too much after I have done this. Now Rosetta Script offers this nice functionality of <SimpleMetrics> where you can specify all kind of metrics that should be evaluated for the generated poses.
So in order to do this afterwards without adding additional overhead to an barely already running script we can simply write a new rescore.xml where we insert all the metrics our heart desires
Here a PDB consisting of 3 chains A,B and C is to be rescored. The Protein here was a Polymerase with DNA bound, which is
the reaseon for the different chains: The protein is chain A and the two strains of the DNA are labeled as chains B and C.
Therefore we need multiple entries in the <RESIDUE_SELECTORS> region to do things with these regions.
Initially I wanted to calculate the interface energy not only between the DNA and Protein but also between the Protein and the single DNA strains. However it seems you can’t reuse a simple metric. At least I always got parsing errors from the Rosetta XML script verification stage.
Only upon removing all but one of the identical Simple_Metrics, would my script run.
To rescore the previously generated poses we need to generate a pdb_list as input, which is simply a text file containing the path
to all the pdb poses. To run the rescoring it is convenient to write all the flags into a single file rosetta_rescore.options.
Using this we can then run the rescoring directly by doing
mpirun –np $N_CORES rosetta_scripts.mpi.linuxgccrelease @rosetta_rescore.options
As I did my run on the LB2 Cluster at TU-Darmstadt, I wrote a slurm script for doing this which looks as follows.
And there you have it, this is nothing special however I often enough forget how to do it and therefore thought
it could be interesting to have this documented somewhere, where it eventually might also help someone else.